Computer Aided Drug Discovery (CADD)
Our work in computer-aided drug discovery focuses on the development of reliable, physically grounded, and automated computational approaches for predicting molecular recognition events. The primary emphasis lies on free energy perturbation (FEP) methods, workflow automation, and the integration of machine learning with simulation protocols to enhance predictive performance and efficiency.
Free Energy Perturbation Methods
We have developed two complementary FEP workflows within the molecular dynamics software Q:
- QligFEP – an automated workflow for calculating relative binding free energies of small molecules to protein targets. It supports high-throughput ligand series analysis and delivers reproducible ΔΔG estimates that correlate with experiment.
- QresFEP – a workflow for computing the effect of protein mutations on ligand binding or structural stability, enabling quantitative analysis of receptor variants and resistance mechanisms.
QligFEP and QresFEP together provide a unified framework for studying both ligand and receptor perturbations in a thermodynamically rigorous manner.
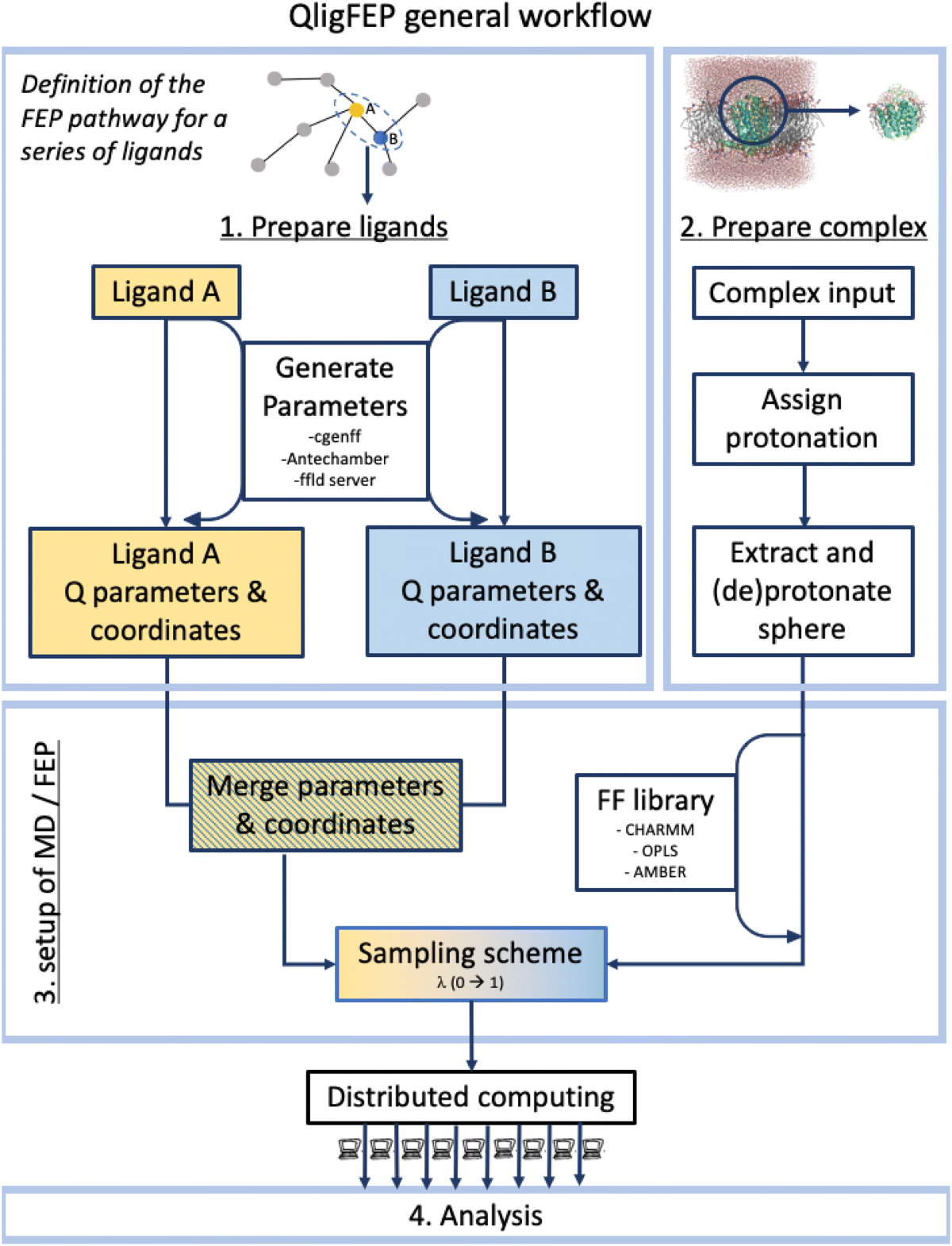
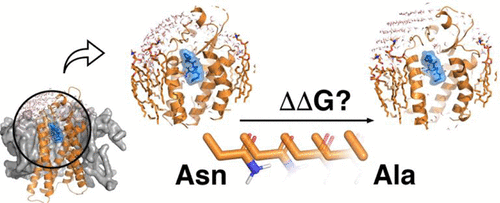
Machine Learning Enhanced FEP Workflows
We are developing approaches that combine free energy simulations with machine learning (ML) to improve the efficiency, convergence, and scope of FEP-based predictions. ML methods are applied to select representative compounds for simulation, optimise alchemical transformation pathways, and predict convergence behaviour. This integration enables active learning cycles where physics-based and data-driven models reinforce each other.

Applications
These computational methodologies are applied to a wide range of biochemical systems, including G protein-coupled receptors (GPCRs), kinases, and other therapeutically relevant targets. By combining automated FEP calculations, mutation analysis, and ML-guided prioritisation, our lab contributes to structure-based ligand design, affinity optimisation, and mechanistic interpretation of receptor function.